La distrofia miotónica de tipo 1 es una miopatía hereditaria autosómica dominante que se caracteriza por mostrar una afectación multisistémica, con dos formas de presentación típicas en el adulto y otra congénita. El fenotipo de presentación varía con distinto grado de afectación sistémica y muscular, y esta última es más típica de la forma adulta [1,2]. El diagnóstico de certeza se realiza mediante un test que detecte la afectación del gen DMPK [2], aunque podemos establecer un diagnóstico de sospecha a través de la sintomatología clásica neuromuscular y multisistémica, incluyendo el sistema nervioso central, para lo cual la resonancia magnética muestra hallazgos de alta sospecha en un adecuado contexto clínico.
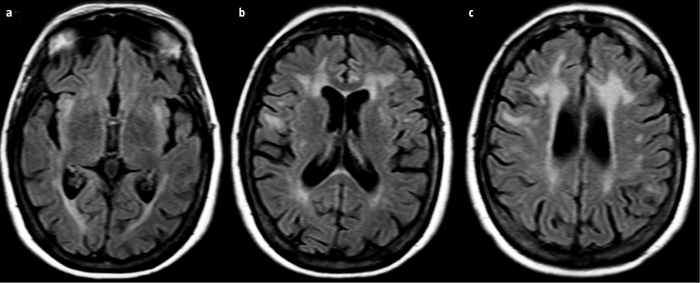
Caso clínico. Varón de 41 años, con sospecha clínica de distrofia neuromuscular, basada en la presencia de atrofia de musculatura cervicofacial y debilidad en las manos, junto con alopecia temprana sin antecedentes familiares. Se realizó una resonancia magnética craneal que presentó lesiones confluentes hiperintensas en T2-TSE y T2-FLAIR, hipointensas en T1 y con aumento de la difusión en DWI (hiperintensidad en DWI y mapa ADC) en la sustancia blanca profunda subcortical en ambos lóbulos temporales (Fig. 1), así como frontales y periatriales bilaterales y con ligero adelgazamiento del cuerpo calloso, y leve atrofia cerebral generalizada (Fig. 2). Estos hallazgos se consideraron característicos de la distrofia miotónica de tipo 1 y el paciente se sometió a un test genético que confirmó la enfermedad.
Figura 1. a) Axial T2-TSE; b) Axial T2-FLAIR; c) Mapa ADC de secuencia de difusión; d) Coronal T1-IR. Se aprecia hiperintensidad en las secuencias T2 y FLAIR e hipointensidad en T1 y sin restricción de la difusión en la sustancia blanca subcortical del polo anterior de ambos lóbulos temporales, con leve atrofia cerebral generalizada.

Figura 2. Axial T2-FLAIR del tercer ventrículo (a), cabeza del caudado (b) y centros semiovales (c). Se aprecian múltiples lesiones hiperintensas en T2-FLAIR en la sustancia blanca periventricular de ambos lóbulos temporales, frontales y periatrial bilateral, así como leve atrofia cerebral generalizada con dilatación pasiva secundaria del sistema ventricular.
Discusión. La distrofia miotónica de tipo 1 es una miopatía hereditaria producida por la alteración del gen DMPK con repetición del trinucleótido CGT más de 34 veces [2], que puede mostrar distinto fenotipo en función de la forma de presentación. La forma clásica del adulto presenta típicamente debilidad y atrofia muscular con afectación predominante de la musculatura distal y miotonía, mientras que la forma media del adulto muestra menor grado de afectación muscular, y la congénita, mayor afectación multisistémica e intelectual [1,2].
La forma clásica del adulto presenta una alteración de la sustancia blanca cerebral profunda periventricular, con lesiones hiperintensas en secuencias potenciadas en T2 sin restricción de la difusión, característicamente de las astas frontales y temporales, con prominencia de los espacios perivasculares de Virchow-Robin [3] y diferente grado de atrofia del cuerpo calloso y la sustancia gris cortical, talámica y de los ganglios basales [4]; por tanto, con la resonancia magnética craneal se dispone de un método no invasivo para el primer abordaje y establecer el diagnóstico de sospecha en pacientes con clínica sugestiva de distrofia miotónica.
Bibliografía
↵ 1. Grupo de Estudio de Enfermedades Neuromusculares de la Sociedad Española de Neurología. Algoritmos diagnósticos en las miopatías hereditarias. URL: http://genm.sen.es. [22.04.2018].
↵ 2. Bird TD. Myotonic dystrophy type 1. Synonym: Steinert’s disease. Genes Review 2015. URL: https://www.ncbi.nlm.nih.gov/books/NBK1165/. [22.04.2018].
↵ 3. Jakkani R, Jyoti S, Ahmed M, Thomas MM. Magnetic resonance imaging findings in adult-form myotonic dystrophy type 1. Singapore Med J 2012; 53: e150-2.
↵ 4. Minnerop M, Weber B, Schoene-Bake JC, Roeske S, Mirbach S, Anspach C, et al. The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain 2011; 134: 3527-43.
© 2019 Revista de Neurología
 OPEN ACCESS
OPEN ACCESS

