TEXTO COMPLETO(solo disponible en lengua castellana / Only available in Spanish)
Las mutaciones del gen CASK se pueden presentar clínicamente como microcefalia con hipoplasia pontocerebelosa o como discapacidad intelectual ligada al cromosoma X, con o sin nistagmo [1]. Estas mutaciones producen fenotipos distintos en función del sexo. Las manifestaciones en los varones no están tan bien definidas como en las mujeres porque existen pocos casos descritos debido a la alta letalidad en edades tempranas, incluso intraútero [2], pero coinciden en los rasgos principales: microcefalia e hipoplasia pontocerebelosa en resonancia magnética. Se presenta el caso de un niño de 9 años con microcefalia con hipoplasia pontocerebelosa, con una mutación de tipo missense en hemicigosis del gen CASK no descrita previamente.
Varón, nacido de parto eutócico, único hijo de padres sanos no consanguíneos. Antecedentes perinatales sin interés. En el nacimiento, destacaba fenotipo ligeramente peculiar con perímetro cefálico en el percentil 3, orejas despegadas de implantación baja y macroftalmia.
A los 12 meses inició crisis pluricotidianas, polimorfas, inicialmente mioclónicas en salvas, y posteriormente crisis focales motoras, tónicas y atónicas. Los registros de electroencefalograma seriados fueron compatibles con una encefalopatía epiléptica grave. A los 5 años, los electroencefalogramas mostraban actividad de fondo globalmente lentificada y un patrón en el sueño con actividad epileptiforme difusa, bilateral y multifocal prácticamente continua. Recibió tratamiento con numerosos fármacos antiepilépticos en distintas combinaciones y con dieta cetógena, pero no se alcanzó el control completo de las crisis en ningún momento.
Se realizaron las siguientes pruebas complementarias: CGH-array, sin alteraciones. Estudio metabólico: aminoácidos en sangre y orina, ácidos orgánicos en orina, ácido pipecólico en plasma, y neurotransmisores, láctico y pirúvico en líquido cefalorraquídeo, con resultados dentro de la normalidad. Biopsia muscular con estudio anatomopatológico y estudio de cadena respiratoria, normales.
También se realizó un estudio enzimático en fibroblastos que descartó gangliosidosis GM1 y GM2, y ceroidolipofuscinosis de los tipos 1 y 2.
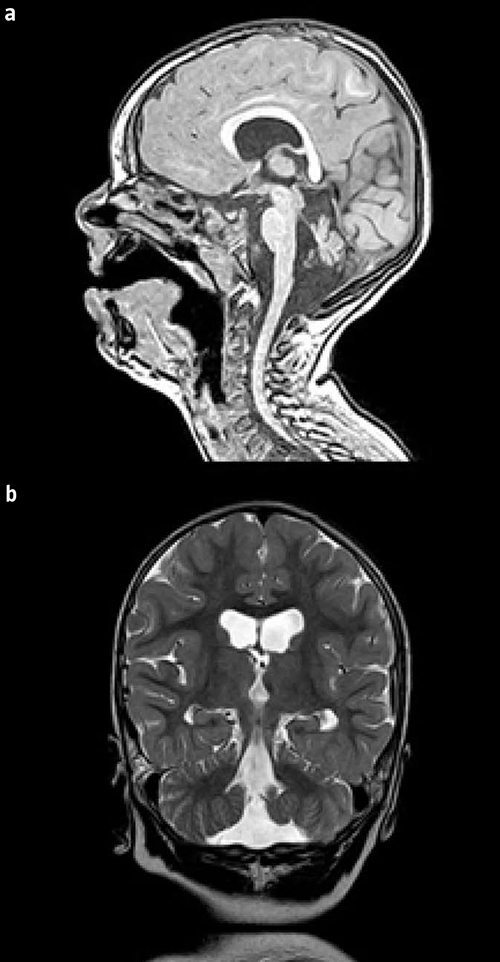
La resonancia magnética cerebral efectuada en el período neonatal no mostró alteraciones relevantes. A los 2 años de edad, la resonancia magnética mostró atrofia cerebelosa importante con mayor afectación del vermis (Figura). En el panel de encefalopatías epilépticas se detectó mutación missense c.532G>A (p.Gly178Arg) en el gen CASK en hemicigosis de novo. La mutación era de cambio de sentido y afectaba a dominios conservados de la proteína. Aunque no se ha descrito previamente, los estudios de predicción bioinformáticos realizados consideraron que se trataba de una variante patógena.
Figura. Imagen en T1 sagital (a) y en T2 coronal (b) a los 2 años de edad que muestra atrofia cerebelosa de predominio en el vermis.
Durante el seguimiento, el paciente ha desarrollado una microcefalia progresiva hasta –5,83 desviaciones estándares, retraso psicomotor grave, hipotonía axial con hipertonía apendicular y nistagmo horizontal-rotatorio de predominio en la mirada hacia la derecha. Con 9 años, no presenta control cefálico ni claro contacto visual. No tiene lenguaje oral y la comprensión verbal está muy afectada. Precisa desplazarse en silla de ruedas y es dependiente para todas las actividades de la vida diaria.
El síndrome de microcefalia con hipoplasia pontocerebelosa en los varones es mucho menos frecuente que en las mujeres, y se debe, en la mayoría de las ocasiones, a mutaciones de pérdida de función en hemicigosis en el gen CASK [3]. Este gen codifica una proteína que interacciona con factores de transcripción importantes durante el desarrollo neuronal [2]. En nuestro caso, la mutación es de tipo missense, la cual se ha notificado tan sólo en tres casos de 41 pacientes con microcefalia con hipoplasia pontocerebelosa [3], y ninguna de ellas coincide con la mutación de nuestro paciente. En los varones, el síndrome de microcefalia con hipoplasia pontocerebelosa presenta un fenotipo más grave que en las mujeres, y se caracteriza por retraso psicomotor grave, microcefalia leve o moderada, y epilepsia precoz y refractaria. Se han descrito casos de síndrome de West [4], síndrome de Ohtahara [5] y epilepsia mioclónica [6].
En este síndrome, la resonancia magnética muestra grados variables de hipoplasia pontocerebelosa y en el mesencéfalo, con un tamaño normal del cuerpo calloso, lo que da la impresión de su engrosamiento. Esta imagen puede ser una pista para detectar pacientes con la mutación en el gen CASK [7]. Cabe destacar que este paciente tenía una primera resonancia magnética sin alteraciones y ha desarrollado posteriormente atrofia cerebelosa, lo cual no se ha descrito en esta patología. Además, hasta donde conocemos, no se ha informado de casos de pacientes con microcefalia con hipoplasia pontocerebelosa y mutación en el gen CASK en España.
La variabilidad fenotípica y de las mutaciones del gen CASK puede explicar los distintos hallazgos radiológicos de los casos clínicos notificados hasta ahora. No se ha podido demostrar que exista una correlación entre la clínica y la gravedad de las lesiones en la resonancia magnética [7,8]. Este caso destaca por tratarse de una mutación missense, no descrita hasta la fecha, en un niño que actualmente tiene 9 años, a pesar de la letalidad de este síndrome en los varones.
Creemos que la búsqueda de mutaciones en el gen CASK debería realizarse en pacientes que presenten características clínicas compatibles con microcefalia, retraso mental e hipoplasia pontocerebelosa.
Bibliografía
↵1. Moog U, Uyanik G, Kutsche K. CASK-related disorders. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al, eds. GeneReviews. Seattle, WA: University of Washington; 2013.
↵2. Najm J, Horn D, Wimplinger I, Golden JA, Chizhikov VV, Sudi J, et al. Mutations of CASK cause an X- linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat Genet 2008; 40: 1065-7.
↵3. Hayashi S, Uehara D, Tanimoto K, Mizuno S, Chinen Y, Fukumura S, et al. Comprehensive investigation of CASK mutations and other genetic etiologies in 41 patients with intellectual disability and microcephaly with pontine and cerebellar hypoplasia (MICPCH). PLoS One 2017; 12: e0181791.
↵4. Takanashi J, Okamoto N, Yamamoto Y, Hayashi S, Arai H, Takahashi Y, et al. Clinical and radiological features of Japanese patients with a severe phenotype due to CASK mutations. Am J Med Genet 2012; 158A: 3112-8.
↵5. Saitsu H, Kato M, Osaka H, Moriyama N, Horita H, Nishiyama K, et al. CASK aberrations in male patients with Ohtahara syndrome and cerebellar hypoplasia. Epilepsia 2012; 53: 1441-9.
↵6. Nakamura K, Nishiyama K, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, et al. De novo CASK mutation in pontocerebellar hypoplasia type 3 with early myoclonic epilepsy and tetralogy of Fallot. Brain Dev 2014; 36: 272-3.
↵7. Takanashi J, Arai H, Nabatame S, Hirai S, Hayashi S, Inazawa J, et al. Neuroradiologic features of CASK mutations. AJNR Am J Neuroradiol 2010; 31: 1619-22.
↵8. Burglen L, Chantot-Bastaraud S, Garel C, Milh M, Touraine R, Zanni G, et al. Spectrum of pontocerebellar hypoplasia in 13 girls and boys with CASK mutations: confirmation of a recognizable phenotype and first description of a male mosaic patient. Orphanet J Rare Dis 2012; 7: 18.
Si ya es un usuario registrado en Neurologia, introduzca sus datos de inicio de sesión.
Rellene los campos para registrarse en Neurologia.com y acceder a todos nuestros artículos de forma gratuita
¿Olvidó su contraseña? Introduzca su correo electrónico y le haremos llegar una nueva
Estimado usuario de Revista de Neurología,
Debido a la reciente fusión por absorción de VIGUERA EDITORES, S.L.U., la entidad gestora de las publicaciones de Viguera Editores, entre ellas, Revista de Neurología, por EVIDENZE HEALTH ESPAÑA, S.L.U., una de las sociedades también pertenecientes al Grupo Evidenze, y con la finalidad de que Usted pueda seguir disfrutando de los contenidos y distintos boletines a los que está suscrito en la página web de neurologia.com, es imprescindible que revise la nueva política de privacidad y nos confirme la autorización de la cesión de sus datos.
Lamentamos informarle que en caso de no disponer de su consentimiento, a partir del día 28 de octubre no podrá acceder a la web de neurologia.com
Para dar su consentimiento a seguir recibiendo la revista y los boletines de neurologia.com vía correo electrónico y confirmar la aceptación de la nueva política de privacidad, así como la cesión de sus datos a Evidenze Health España S.L.U., el resto de las entidades del Grupo Evidenze y sus partners y colaboradores comerciales, incluyendo la posibilidad de llevar a cabo transferencias internacionales a colaboradores extranjeros, pulse en el siguiente enlace:
 OPEN ACCESS
OPEN ACCESS