La ataxia episódica tipo 2 (AE2; MIM #108500) es una enfermedad genética de inicio infantil con herencia autosómica dominante por variantes patógenas en el gen CACNA1A (*601011; 19p13). Este gen codifica para la subunidad α-1A de los canales de calcio dependientes del voltaje de las células de Purkinje cerebelosas [1]. Aunque es la forma más frecuente de ataxia episódica familiar, la prevalencia es baja (<1/100.000) [2]. La sintomatología puede solaparse con otras causas de vértigo recurrente [3]. Clínicamente cursa con episodios de ataxia cerebelosa de horas de duración, habitualmente precedidos de desencadenantes (estrés, calor, ejercicio...). Entre los episodios son frecuentes las manifestaciones de disfunción cerebelosa, como la ataxia o el nistagmo evocado por la mirada [4,5]. La migraña (hemipléjica o no), la epilepsia, la distonía infantil episódica y las alteraciones neuropsiquiátricas son síntomas asociados a CACNA1A [1,6,7]. Es característica la respuesta a la acetazolamida y la 4-aminopiridina [3]. El objetivo de este estudio es presentar una serie de pacientes con diagnóstico molecular de AE2, y describir sus características epidemiológicas, genéticas, clínicas, radiológicas y terapéuticas.
Material y métodos
Se realizó una revisión retrospectiva de pacientes con diagnóstico molecular de AE2, seguidos entre 1988 y 2022 en el Servicio de Neurología de nuestro centro (base de datos de la Unidad de Trastornos del Movimiento).
Los estudios genéticos de los probandos de las familias 1, 2 y 4 se realizaron mediante secuenciación de Sanger del gen completo CACNA1A (NM_001127221.2). El probando de la familia 3 se analizó mediante secuenciación de exoma y posterior filtrado de un panel virtual de 70 genes de encefalopatías epilépticas (incluyendo CACNA1A). En el probando de la familia 5 se realizó un panel de secuenciación de nueva generación de 62 genes asociados a ataxias. Todos los estudios de segregación de las variantes detectadas en el resto de los familiares se llevaron a cabo por secuenciación de Sanger.
Los estudios genéticos y las variantes descritas se realizaron o revisaron por el Servicio de Genética de nuestro hospital. Las variantes en CACNA1A se clasificaron como patógenas o probablemente patógenas de acuerdo con los criterios establecidos en 2015 por el American College of Medical Genetics and Genomics a partir de la información extraída de bases de datos de variantes genéticas (HGMD y ClinVar) o bases de datos poblacionales (gnomAD), y de herramientas de clasificación semiautomática y de predicción in silico disponibles en el momento de su revisión.
Las imágenes radiológicas fueron revisadas por un radiólogo especialista en neurorradiología. El análisis de datos se basó en estadística descriptiva.
Resultados
Aspectos epidemiológicos y genéticos
Se incluye a 10 pacientes, procedentes de cinco familias no relacionadas (seis mujeres y cuatro varones) (Fig. 1). La mediana de edad de inicio de los síntomas fue de 10 años, y la mediana de edad en el momento del diagnóstico, de 37,5 años. Con ello, se obtuvo una mediana de retraso diagnóstico desde el inicio sintomático de 20 años (Fig. 2). En el ámbito molecular, se detectaron cinco variantes genéticas (una variante de cambio de sentido o missense; tres variantes sin sentido o stop gain; y una de desplazamiento del marco de lectura o frameshift) en CACNA1A (Tabla I). Dos pacientes (familia 3) fueron una pareja consanguínea heterocigota que asoció mortalidad infantil prematura por encefalopatía epiléptica de inicio precoz de tipo 42 en su descendencia homocigota.
Figura 1. Genealogía de las cinco familias de la serie. En negro, pacientes diagnosticados de ataxia episódica tipo 2 con confirmación molecular (heterocigotos para CACNA1A), salvo los englobados con las siglas EEIP42 (encefalopatía epiléptica de inicio precoz de tipo 42, homocigotos para CACNA1A); en gris, familiares que asociaban sintomatología relacionada con CACNA1A sin ataxia episódica tipo 2; en blanco, pacientes libres de síntomas; y marcados con asterisco, sujetos de nuestro estudio. TLP: trastorno límite de la personalidad.
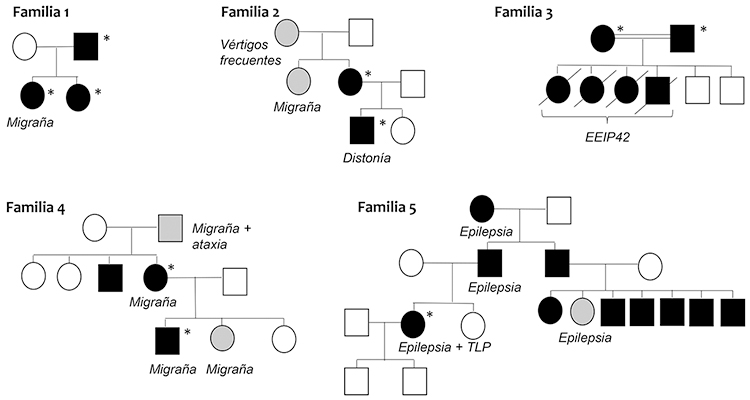
Figura 2. Se representa, para cada paciente, la edad de inicio de los síntomas (flecha gruesa) y la edad en el momento del diagnóstico (flecha fina). La barra muestra el período libre de síntomas (negro) y el retraso diagnóstico (gris). La paciente 1 tuvo un inicio en la infancia a edad no determinada, por lo que no se refleja en la gráfica.
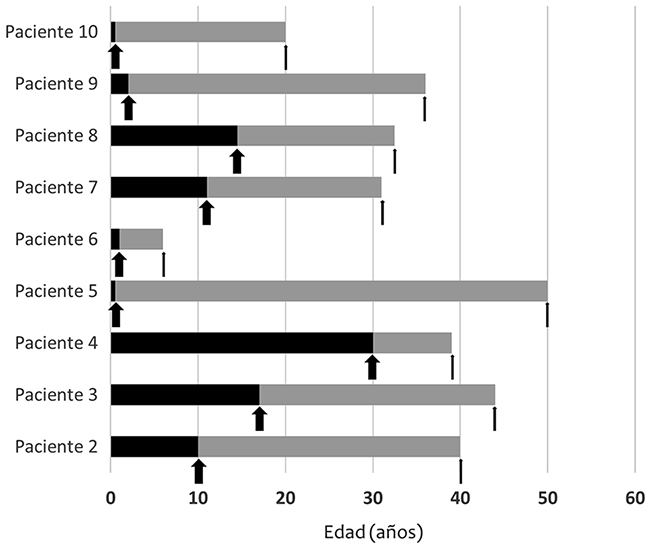
Tabla I. Variantes en el gen CACNA1A confirmadas en nuestra serie.
|
|
Familia
|
Variantes
|
N.o de pacientes
|
Clasificación
|
Descrita
|
|
1
|
c.5569C>T; p.(Arg1857*)
|
3
|
Patogénica
|
Sí
|
|
2
|
c.6655C>T; p.(Pro2222Leu)
|
2
|
Patogénica
|
Sí
|
|
3
|
c.2767C>T; p.(Arg923*)
|
2
|
Probablemente patogénica
|
Sí
|
|
4
|
c.5547T>A; p.(Tyr1849*)
|
2
|
Patogénica
|
Sí
|
|
5
|
c.5588_5589del; p.(Leu1863Argfs*9)
|
1
|
Probablemente patogénica
|
No
|
Aspectos clínicos
La frecuencia episódica inicial fue semanal en el 50% de los pacientes. El 80% de los pacientes tuvo desencadenantes de los episodios, y el estrés fue el más habitual. Seis pacientes presentaron otros desencadenantes (café, ejercicio, alcohol o calor). La duración de los episodios fue menor de 24 horas en el 70%, y se resolvieron en todos con el decúbito o el sueño. La sintomatología episódica subjetiva percibida por los pacientes era inestabilidad (100%) más que síndrome vertiginoso (20%) o disartria (10%). Seis pacientes (60%) presentaron ataxia interepisódica, que fue sutil o leve en la mayoría (cinco de seis). La paciente con una mayor frecuencia de episodios (diaria) presentaba una ataxia interepisódica moderada (deambulación con bastón). El 70% presentó nistagmo interepisódico evocado por la mirada, más frecuente en pacientes con mayor frecuencia episódica. El 50% de los pacientes presentó otros síntomas asociados a CACNA1A (Fig. 1): tres, migraña no hemipléjica; uno, epilepsia; uno, distonía interepisódica infantil; y uno, trastorno neuropsiquiátrico (trastorno límite de la personalidad). No detectamos casos de migraña hemipléjica familiar. De los 10 pacientes, siete tenían historia familiar de ataxia episódica; cuatro, de migraña; y tres, de epilepsia (Tabla II).
Tabla II. Variables clínicas, radiológicas y terapéuticas de cada paciente de la serie.
|
|
Paciente
|
Edad inicial (años) y sexo
|
Edad en el momento del diagnóstico (años)
|
Frecuencia inicial de los episodios
|
Desencadenantes
|
AI
|
NIEM
|
Síntomas CACNA1A asociados
|
AC
|
Respuesta a la ACZ/efectos adversos
|
|
1
|
Indeterminada/mujer
|
45
|
Semanal
|
Estrés
|
Leve
|
Sí
|
No
|
V+H
|
Sí/litiasis
|
|
2
|
10/mujer
|
40
|
Semanal
|
Estrés, café
|
No
|
Sí
|
Epilepsia, TLP
|
No
|
Sí/litiasis y vómitos
|
|
3
|
17/varón
|
44
|
Anual
|
Indeterminados
|
Sutil
|
No
|
No
|
–
|
–
|
|
4
|
30/mujer
|
39
|
Esporádica
|
Indeterminados
|
Sutil
|
No
|
No
|
–
|
–
|
|
5
|
0,5/mujer
|
50
|
Diaria
|
Estrés
|
Moderada
|
Sí
|
Migraña
|
V
|
Sí/no
|
|
6
|
1/varón
|
6
|
Semanal
|
Ejercicio
|
No
|
No
|
Distonía infantil episódica
|
No
|
Sí/no
|
|
7
|
11/varón
|
31
|
Anual
|
Estrés, calor
|
No
|
Sí
|
No
|
V + H
|
No (dosis bajas)/mareos
|
|
8
|
14/mujer
|
32
|
Semanal
|
Estrés, café
|
No
|
Sí
|
No
|
No
|
Sí/litiasis
|
|
9
|
2/mujer
|
36
|
Semanal
|
Estrés
|
Sutil
|
Sí
|
Migraña
|
No
|
Sí/no
|
|
10
|
0,5/varón
|
20
|
Mensual
|
Estrés, alcohol
|
Leve
|
Sí
|
Migraña
|
H
|
No (dosis bajas)/mareos
|
AC: atrofia cerebelosa; ACZ: acetazolamida; AI: ataxia interepisódica; H: atrofia cerebelosa hemisférica (uni- o bilateral); NIEM: nistagmo interepisódico evocado por la mirada; TLP: trastorno límite de la personalidad; V: atrofia cerebelosa vermiana.
|
Aspectos radiológicos
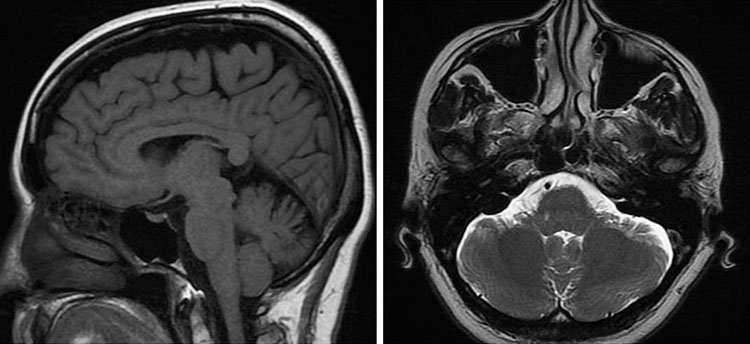
Se realizó un estudio de neuroimagen en ocho pacientes (Fig. 3). Cuatro presentaron atrofia cerebelosa (edad media: 53 años), vermiana o hemisférica, uni- o bilateral. Todos los pacientes con mayor grado de ataxia interepisódica (leve o moderado) presentaban atrofia cerebelosa, mientras que sólo el 75% de los pacientes con atrofia cerebelosa asoció ataxia interepisódica. Todos los pacientes con atrofia cerebelosa presentaron nistagmo interepisódico evocado por la mirada.
Figura 3. Resonancia magnética de la paciente 1 que muestra atrofia cerebelosa (izquierda, T1 sagital; derecha, T2 axial).
Aspectos terapéuticos
Ocho pacientes iniciaron acetazolamida en pauta permanente durante el seguimiento (mediana de tratamiento: 12 años). El 75% presentó respuesta terapéutica a los síntomas episódicos con dosis iguales o superiores a 375 mg/día, y la frecuencia disminuyó en cinco (de semanal a mensual) y la intensidad en uno. Dos pacientes no presentaron respuesta terapéutica a la acetazolamida, aunque sólo alcanzaron dosis bajas de acetazolamida (250 mg/día) por mala tolerancia. La acetazolamida no modificó la ataxia ni el nistagmo evocado por la mirada interepisódicos durante el seguimiento. Los efectos adversos se presentaron en el 63% de los pacientes tratados con acetazolamida, y el más frecuente fue la nefrolitiasis. Dos pacientes asociaron mareos e intolerancia a la acetazolamida, lo que motivó su suspensión, y un paciente presentó vómitos, lo que requirió un ajuste de la dosis. Uno de los casos precisó nefrostomía con catéter doble J, lo que demandó suspender la acetazolamida e iniciar 4-aminopiridina. Tras este cambio, se obtuvo una respuesta total.
Discusión
La AE2 suele tener un inicio infantojuvenil antes de los 20 años [4] (el 90% en nuestra serie). Se han descrito también casos de inicio tardío [2,6]. El diagnóstico requiere un elevado índice de sospecha, dado que la sintomatología episódica puede solaparse con otras causas de vértigo recurrente [3]. El retraso diagnóstico está poco documentado en la bibliografía, pero es habitual, dada la difícil caracterización del cuadro clínico, bien por pacientes, bien por médicos no neurólogos. La mediana de retraso diagnóstico en nuestra serie fue de 20 años (Fig. 2), similar a la serie de Mantuano et al [7] y al caso notificado por Muro-García et al [8].
Los pacientes con AE2 pueden asociar a los síntomas episódicos signos y síntomas interepisódicos, como el nistagmo evocado por la mirada o la ataxia persistente [2,4]. La relación entre frecuencia de episodios y manifestaciones interepisódicas fue documentada por Nachbauer et al [6], y es sabido que la ataxia puede ser progresiva durante el curso de la enfermedad [1,4]. Nuestra serie sugiere también una posible asociación entre una mayor frecuencia episódica, y el desarrollo de ataxia interepisódica y de nistagmo interepisódico evocado por la mirada, basado en que la paciente con mayor frecuencia de episodios tenía el mayor grado de ataxia interepisódica.
El estudio de los síntomas asociados a CACNA1A está cobrando relevancia tanto en su aspecto diagnóstico como en su manejo clínico [1]. La migraña (hemipléjica o no) es el síntoma asociado a AE2 más frecuente [5], aunque la epilepsia, las alteraciones neuropsiquiátricas o la distonía interepisódica infantil son habituales. En nuestra serie, dicha sintomatología presentó gran variabilidad intrafamiliar, pero la migraña y la epilepsia tendieron a agruparse por familias (Fig. 1). En este sentido, cabe destacar que se llegó al diagnóstico de AE2 en la familia 3 sólo tras varios casos de mortalidad infantil prematura por encefalopatía epiléptica de inicio precoz de tipo 42 en su descendencia homocigota [9].
La atrofia cerebelosa es un hallazgo común en los pacientes con AE2, especialmente los que tienen una ataxia interepisódica más grave [2,5]. Mantuano et al [7] reflejan que el 27,3% de los pacientes con confirmación molecular presenta atrofia cerebelosa, menos que el 50% de nuestra serie. Es relevante comentar que, en contra de lo aparentemente esperable, ni en nuestro estudio ni en la bibliografía hemos encontrado asociación entre frecuencia episódica o tiempo de evolución y desarrollo de atrofia cerebelosa.
Los síntomas episódicos de AE2 se caracterizan por una buena respuesta a la acetazolamida y la 4-aminopiridina. La acetazolamida es eficaz en un 50-75% de los pacientes con AE2 [2,3,7] en dosis entre 250 y 1.000 mg diarios. Sin embargo, es frecuente su retirada debido a efectos adversos, de los cuales la nefrolitiasis es el más destacable [3]. Según nuestros resultados, como también comenta Baloh [5], hay una posible respuesta dependiente de la dosis a la acetazolamida (mayor eficacia a partir de 375 mg/día), aunque a expensas de aumentar los efectos adversos. Nuestra experiencia con la 4-aminopiridina es de sólo una paciente, pero, como sugieren Strupp et al [10], su efectividad parece ser mayor que la de la acetazolamida. Su escasa disponibilidad y elevado precio pueden ser una barrera para su uso.
Nuestro estudio presenta ciertas limitaciones, ya que se trata de una serie no aleatorizada, retrospectiva y descriptiva, con un tamaño muestral reducido. En este sentido, puede llamar la atención el pequeño número de pacientes recogido, incluso siendo nuestro hospital un centro de referencia, pero el trabajo no debe considerarse un estudio epidemiológico y, además, la AE2 es una enfermedad con muy baja prevalencia [2].
Conclusiones
En nuestra serie, la AE2 presentó una alta variabilidad inter- e intrafamiliar respecto a la frecuencia episódica y a la asociación de otras manifestaciones relacionadas con CACNA1A. El fenotipo más frecuente fueron pacientes con episodios de inestabilidad de horas de duración, semanales, con estrés como desencadenante, ataxia persistente de grado variable y nistagmo evocado por la mirada. La acetazolamida fue eficaz en dosis altas, aunque con efectos adversos, fundamentalmente nefrolitiasis. El retraso diagnóstico fue frecuente, por lo que se requiere un elevado índice de sospecha.
Bibliografía
↵ 1. Indelicato E, Boesch S. From genotype to phenotype: expanding the clinical spectrum of CACNA1A variants in the era of next generation sequencing. Front Neurol 2021; 12: 639994.
↵ 2. Kipfer S, Strupp M. The clinical spectrum of autosomal-dominant episodic ataxias. Mov Disord Clin Pract 2014; 1: 285-90.
↵ 3. Strupp M, Zwergal A, Brandt T. Episodic ataxia type 2. Neurotherapeutics 2007; 4: 267-73.
↵ 4. Jen J, Kim GW, Baloh RW. Clinical spectrum of episodic ataxia type 2. Neurology 2004; 62: 17-22.
↵ 5. Baloh RW. Episodic ataxias 1 and 2. Handb Clin Neurol 2012; 103: 595-602.
↵ 6. Nachbauer W, Nocker M, Karner E, Stankovic I, Unterberger I, Eigentler A, et al. Episodic ataxia type 2: phenotype characteristics of a novel CACNA1A mutation and review of the literature. J Neurol 2014; 261: 983-91.
↵ 7. Mantuano E, Romano S, Veneziano L, Gellera C, Castellotti B, Caimi S, et al. Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J Neurol Sci 2010; 291: 30-6.
↵ 8. Muro García I, Toribio-Díaz ME, Quintas S. Episodic ataxia: a 20-year diagnostic delay. Ataxia episódica: 20 años de retraso diagnóstico. Neurologia (Engl Ed) 2020; 35: 500-1.
↵ 9. Arteche-López A, Álvarez-Mora MI, Sánchez Calvin MT, Lezana Rosales JM, Palma Milla C, Gómez Rodríguez MJ, et al. Biallelic variants in genes previously associated with dominant inheritance: CACNA1A, RET and SLC20A2. Eur J Hum Genet 2021; 29: 1520-6.
↵ 10. Strupp M, Kalla R, Claassen J, Adrion C, Mansmann U, Klopstock T, et al. A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology 2011; 77: 269-75.
Episodic ataxia type 2: a clinical, genetic and radiological study of 10 patients
Objectives. To describe a series of patients with episodic ataxia type 2 (EA2), attending to epidemiological, clinical, radiological, and therapeutic variables.
Material and methods. Retrospective revision of patients with molecular diagnosis of EA2 (CACNA1A mutations), between 1988 and 2022. Information achieved from the database of our Movement Disorders clinic. A descriptive statistical analysis was made.
Results. Ten patients from five families were analyzed (six women). Median age at diagnosis was 37.5 years-old, with a median diagnostic delay of 20 years. 70% reported familial history of CACNA1A associated symptoms, although 50% presented migraine, epilepsy, dystonia, or neuropsychiatric alterations. Two heterozygous consanguineous patients had homozygotic descendance with infant mortality due to early-onset epileptic encephalopathy type 42. Five pathogenic/probably pathogenic CACNA1A variants were detected. 80% of patients had episodic triggers, being stress the most common. Episodes had a weekly frequency before treatment initiation. Six patients developed chronic ataxia (one patient demand gait support). 50% of patients with neuroimaging presented cerebellar atrophy. Acetazolamide were initiated in 80%, and 75% of them showed improvement of episodic symptoms. Nephrolithiasis was the most frequent side effect.
Conclusions. EA2 has a great intrafamilial and interfamilial phenotypic variability. The most frequent phenotype were weekly episodes of unsteadiness, several hours of length, stress as the main trigger, chronic ataxia and gaze-evoked nystagmus. Acetazolamide is effective, although complications are usual. Neurologist must be alert as diagnostic delay is constant.
Key words. Acetazolamide. CACNA1A. Cerebellar atrophy. Chronic ataxia. Episodic ataxia type 2. Familial hemiplegic migraine.
|
© 2023 Revista de Neurología
 OPEN ACCESS
OPEN ACCESS

