Introducción
BRWD3 es un gen causal conocido de discapacidad intelectual ligada a X. En 2007, variantes de truncamiento en este gen se identificaron en dos familias, con cuatro varones afectos y una mujer, de 250 estudiadas con discapacidad intelectual ligada a X no sindrómica con estudio previo negativo en 62 genes conocidos asociados a discapacidad intelectual ligada a X [1]. Posteriormente se han publicado hasta 57 pacientes [2-7], aunque no todos con su descripción clínica, lo que ha permitido ir definiendo su fenotipo (OMIM 300553, DILX 93). Sus manifestaciones más frecuentes son la discapacidad intelectual, la dismorfia facial, la macrocefalia, el sobrecrecimiento y la obesidad [1-7].
Las variantes más frecuentes son puntuales en el gen, de tipo codón de parada, cambio de marco de lectura, cambio de aminoácido y en los sitios de empalme [1-7]. También se han descrito tres deleciones parciales que incluyen sólo el gen BRWD3 [2,5].
El gen BRWD3 se localiza en Xq21.1, y codifica el bromodominio y dominio de repetición WD, conteniendo la proteína 3, BRWD3, proteína de 1.802 aminoácidos que contiene ocho dominios de repetición WD localizados N-terminales y 2 bromodominios C-terminales. Es un lector epigenético de la acetilación de las histonas que regula la remodelación de la cromatina, la ubiquitinación y la transducción de señales [8]. De forma específica, BRWD3 regula la actividad de KDM5 para equilibrar los niveles de metilación de H3K4 [9]. Se expresa, predominantemente en el período embrionario, en localizaciones múltiples, incluyendo el cerebro, con un papel fundamental en su desarrollo y la plasticidad sináptica [1,5-6].
Presentamos a un varón con una deleción de 586 Kb en la región Xq21.1 que incluye sólo un gen con patología conocida asociada, BRWD3, con fenotipo compatible con los pacientes descritos con variantes patógenas en este gen. Su madre y hermana, asintomáticas, son portadoras de la deleción, que ha aparecido de novo en la madre, ya que la abuela materna no es portadora.
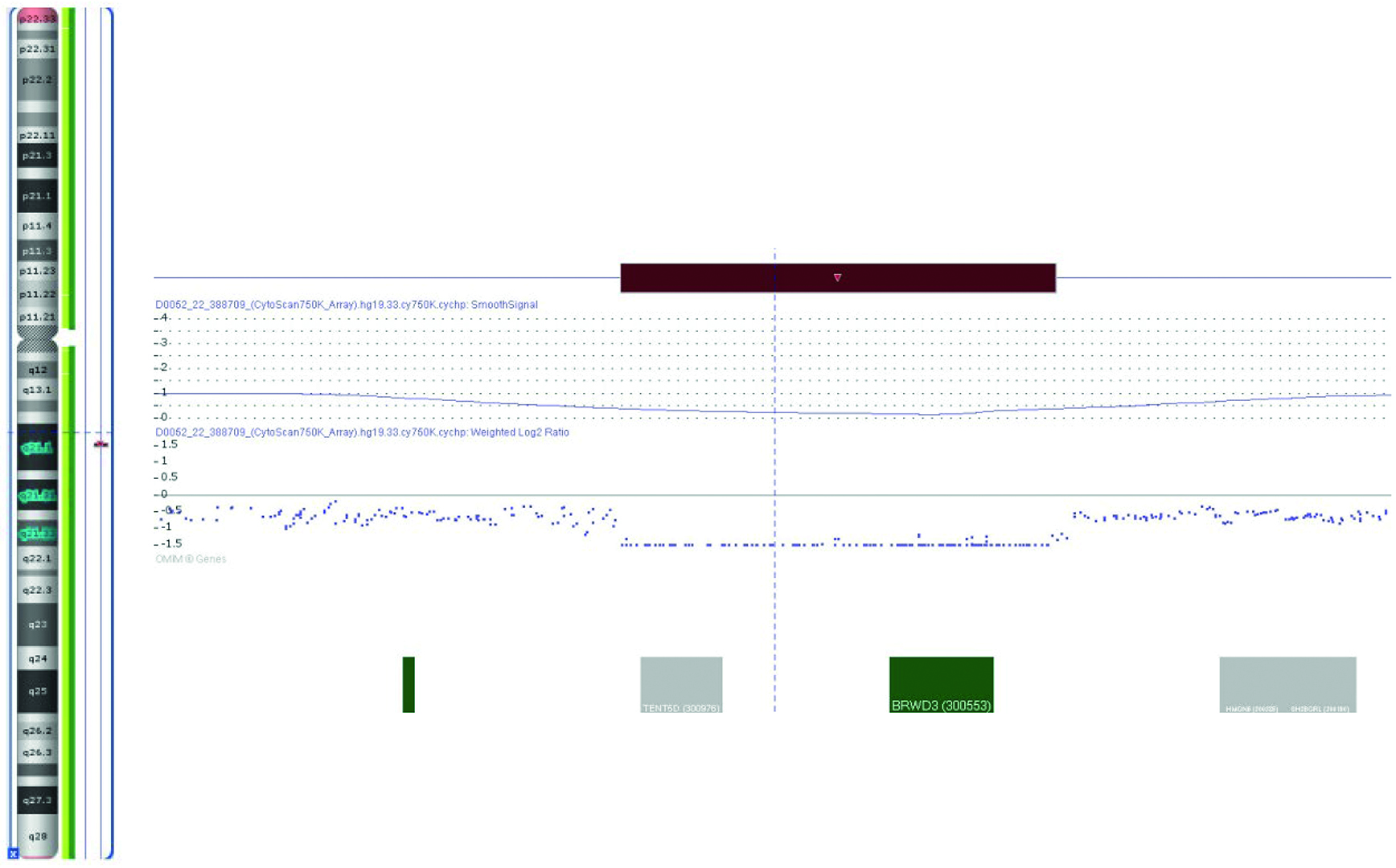
Figura. Array de hibridación genómica comparada del paciente en el que se observa la deleción de 586 kb en la citobanda Xq21.1 arr[GRCh37]Xq21.1(79564583_80150138)x0.
Caso clínico
Varón, hijo de madre de 28 años en el momento de su nacimiento y de padre de 34. Sin consanguinidad, sin historia familiar, nacido de segunda gestación, controlada, de curso normal. Serologías: lúes, toxoplasma, virus de la inmunodeficiencia humana y HBsAg negativos, y rubéola inmune. Cribado combinado del primer trimestre de riesgo bajo. Ecografías prenatales normales. No hay contacto o ingesta conocida de tóxicos y/o teratógenos. Parto eutócico a las 40 semanas. Apgar: 9/10. Peso en el nacimiento: 3.780 g (p50-90, gráficas Fenton). Longitud: 52 cm (p50-90). Perímetro cefálico: 37 cm (p90). Exploración neonatal descrita como normal. Cribados neonatales metabólico y auditivo sin alteraciones.
Estaba en seguimiento por neuropediatría por retraso psicomotor en su evolución: marcha autónoma a los 18 meses y retraso del lenguaje. Asistió a un programa de atención temprana desde el segundo año de vida, con apoyos escolares (audición y lenguaje + pedagogía terapéutica) desde el inicio de la educación infantil y adaptación curricular desde primaria. Cociente intelectual a los 6 años: 68. Actualmente asiste a habilitación funcional. Otros problemas de salud: exotropía alterna descompensada pendiente de cirugía, obesidad en seguimiento por endocrinología infantil y diagnosticado de betatalasemia menor.
Valorado en consulta de genética clínica a los 8,5 años: peso, 45 kg –p95; desviación estándar; (DE) = 1,75–; talla, 138 cm (p77; DE = 0,77); índice de masa corporal, 23,63 (p96; DE = 1,85); y perímetro cefálico, 57 cm (p99; DE = 3,05). Fenotipo con macrocefalia, obesidad troncular con adipomastia, frente amplia, ancha, ojos hundidos, nariz bulbosa, pabellones auriculares despegados ligeramente no displásicos, mentón afilado, úvula corta y ancha, voz nasal, falanges distales de las manos afiladas, pies planos laxos y pene hundido en grasa prepúbica.
Estudio genético. Array SNP/CNV 750K (plataforma GeneChip® System 3000, Affymetrix®, CytoScan Assay P/N 703038 Rev.5. Análisis con programa Chromosome Analysis Suite, Affymetrix® v.3.3): se detecta una deleción de 586 kb en la citobanda Xq21.1 arr[GRCh37]Xq21.1(79564583_80150138)x0 (Figura). La deleción incluye un único gen con patología conocida asociada, BRWD3. Se realiza posteriormente array a la madre y a la hermana, asintomáticas, ambas portadoras de la deleción identificada en el paciente, no presente en la abuela materna.
Discusión
Las manifestaciones clínicas presentes en nuestro paciente (retraso motor y del lenguaje, discapacidad intelectual, macrocefalia, obesidad, dismorfia facial con frente amplia, ojos hundidos, pabellones auriculares despegados y mentón afilado, falanges distales de las manos afiladas y pies planos laxos), con una deleción que incluye únicamente el gen con patología conocida asociada BRWD3, son compatibles con el fenotipo descrito en los casos con variantes patógenas en el gen [1-7].
Las manifestaciones clínicas más frecuentes en los casos con descripción fenotípica de la bibliografía [1-7] (Tabla) son discapacidad intelectual, presente en el 100% de los varones, dismorfia facial (96%), macrocefalia (80%), sobrecrecimiento (57%) y obesidad (44%). El fenotipo neuroconductual incluye en la mayoría de los casos retraso motor y del lenguaje, además de la discapacidad intelectual; también asocia en la mayoría de los casos alteración conductual (64%), incluyendo trastorno por déficit de atención/hiperactividad (22%) y trastorno del espectro autista (17%). El 11% de los pacientes presenta epilepsia. La dismorfia facial se caracteriza por frente amplia prominente, ojos hundidos, pabellones auriculares grandes despegados y mentón prominente. La macrocefalia puede estar presente en el nacimiento, pero en algunos casos se desarrolla posnatalmente. Otras manifestaciones menos frecuentes son dedos largos, alteraciones esqueléticas (escoliosis, cifosis, pectus excavatum), fisura palatina, hipospadias y criptorquidia.
Tabla. Manifestaciones clínicas de los pacientes varones (incluye las referencias 1-7 de la bibliografía, 43 casos en total). Otros pacientes de la bibliografía, hasta 57 en total, no tienen fenotipo descrito o muy parcial.
|
| |
Bibliografía
(43 casos)
|
Nuestro
caso
|
Antropometría al nacimiento
|
|
|
Peso > +2 DE
|
15,45%
|
–
|
Longitud > +2 DE
|
31,6%
|
–
|
Perímetro cefálico > +2 DE
|
60%
|
–
|
Dismorfia facial
|
|
|
Frente amplia prominente
|
72%
|
+
|
Pabellones auriculares grandes despegados
|
53,8%
|
+
|
Mentón prominente
|
42,3%
|
+
|
Otros hallazgos faciales
|
85,2%
|
+
|
Macrocefalia
|
80%
|
+
|
Sobrecrecimiento
|
57,1%
|
–
|
Obesidad
|
44,4%
|
p96
|
Anomalías en las manos y/o los pies
|
53,8%
|
+
|
Otras manifestaciones clínicas
|
86,7%
|
+
|
Neurodesarrollo
|
|
|
Discapacidad intelectual
|
100%
|
+
|
Retraso motor
|
37,5%
|
+
|
Retraso del lenguaje
|
96%
|
+
|
Alteración conductual
|
64%
|
–
|
TDAH
|
22%
|
–
|
TEA
|
17%
|
–
|
Tipo de variante
|
|
|
Codón de parada
|
41,9%
|
|
Cambio de marco de lectura
|
19,3%
|
|
Cambio de aminoácido
|
12,9%
|
|
En el sitio de empalme
|
16,1%
|
|
Deleción que incluye sólo el gen BRWD3
|
9,7%
|
+
|
Madre portadora
|
64%
|
+
|
DE: desviación estándar; TDAH: trastorno por déficit de atención/hiperactividad; TEA: trastorno del espectro autista.
|
Por sus manifestaciones clínicas debemos incluir las alteraciones asociadas a BRWD3 en los síndromes con sobrecrecimiento y discapacidad intelectual, importante para el diagnóstico diferencial.
Sólo se ha descrito a tres mujeres con manifestaciones clínicas en la bibliografía [1,7], el resto de los casos son varones [1-7], y las madres, aparent1as, son portadoras de la variante en un porcentaje elevado de los casos con estudio familiar realizado. De las tres mujeres afectas [1,7], dos de ellas presentan un fenotipo poco expresivo y la tercera [7] tiene una expresividad similar a la de los varones; esta mujer presenta un patrón sesgado completo de inactivación de X a favor del alelo de BRWD3 mutado, lo que posiblemente explica su fenotipo.
Respecto a la correlación genotipo-fenotipo, en 2023 se han publicado [10] los casos de cinco pacientes con epilepsia sin discapacidad intelectual con variantes de cambio de aminoácido en BRWD3 localizadas en los dominios WD repetidos y en los bromodominios, a diferencia de las variantes que originan truncamiento o cambio de aminoácido situadas en otras localizaciones del gen que originan discapacidad intelectual. Junto con los datos previos de evidencia de la expresión de este gen en el cerebro, que es esencial para el neurodesarrollo normal, debemos incluir BRWD3 entre los genes causales de epilepsia.
Conclusiones
Nuestro caso confirma que la haploinsuficiencia debida a la deleción del gen BRWD3 es un mecanismo genético causal de la discapacidad intelectual sindrómica ligada a X asociada al gen BRWD3. Es importante el reconocimiento de esta entidad para su diagnóstico y el seguimiento del paciente, y la realización del estudio familiar para identificar y ofrecer asesoramiento genético a las mujeres de la familia que porten la alteración, puesto que en la gran mayoría de los casos serán asintomáticas, como la madre y la hermana de este paciente.
Bibliografía
↵ 1. Field M, Tarpey PS, Smith R, Edkins S, O’Meara S, Stevens C, et al. Mutations in the BRWD3 gene cause X-linked mental retardation associated with macrocephaly. Am J Hum Genet 2007; 8: 367-74.
↵ 2. Grotto S, Drouin-Garraud V, Ounap K, Puusepp-Benazzouz H, Schuurs-Hoeijmakers J, Le Meur N, et al. Clinical assessment of five patients with BRWD3 mutation at Xq21.1 gives further evidence for mild to moderate intellectual disability and macrocephaly. Eur J Med Genet 2014; 57: 200-6.
↵ 3. Grozeva D, Carss K, Spasic-Boskovic O, Tejada MI, Gecz J, Shaw M, et al. Targeted next-generation sequencing analysis of 1,000 individuals with intellectual disability. Hum Mutat 2015; 36: 1197-204.
↵ 4. Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A, et al. Mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability. Am J Hum Genet 2017; 100: 725-36.
↵ 5. Ostrowski PJ, Zachariou A, Loveday C, Baralle D, Blair E, Douzgou S, et al. Null variants and deletions in BRWD3 cause an X-linked syndrome of mild-moderate intellectual disability, macrocephaly, and obesity: a series of 17 patients. Am J Med Genet C Semin Med Genet 2019; 181: 638-43.
↵ 6. Tenorio J, Alarcón P, Arias P, Ramos FJ, Campistol J, Climent S, et al. MRX93 syndrome (BRWD3 gene): five new patients with novel mutations. Clin Genet 2019; 95: 726-31.
↵ 7. Delanne J, Lecat M, Blackburn PR, Klee EW, Stumpel CTRM, Stegmann S, et al. Further clinical and molecular characterization of an XLID syndrome associated with BRWD3 variants, a gene implicated in the leukemia-related JAK-STAT pathway. Eur J Med Genet 2023; 66: 104670.
↵ 8. Chen WY, Shih HT, Liu KY, Shih ZS, Chen LK, Tsai TH, et al. Intellectual disability-associated dBRWD3 regulates gene expression through inhibition of HIRA/YEM-mediated chromatin deposition of histone H3.3. EMBO Rep 2015; 16: 528-38.
↵ 9. Han D, Schaffner SH, Davies JP, Benton ML, Plate L, Nordman JT. BRWD3 promotes KDM5 degradation to maintain H3K4 methylation levels. Proc Natl Acad Sci U S A 2023; 120: e2305092120.
↵ 10. Tian MQ, Liu XR, Lin SM, Wang J, Luo S, Gao LD, et al. Variants in BRWD3 associated with X-linked partial epilepsy without intellectual disability. CNS Neurosci Ther 2023; 29: 727-35.
X-linked intellectual disability syndrome with macrocephaly due to BRWD3 gene deletion
Introduction. Pathogenic variants in BRWD3 gene have been described as a rare cause of syndromic X-linked intellectual disability. Its phenotype shows neurodevelopmental delay with intellectual disability in all reported patients, facial dysmorphic features, macrocephaly, overgrowth and obesity. The great majority of cases yield point variants in the gene, only three large deletions including only the BRWD3 gene have been reported. The BRWD3 protein is an epigenetic reader that regulates chromatin remodeling. We report a boy with a compatible phenotype and a deletion including only this gene.
Case report. Boy, without family and perinatal pathological background, with neurodevelopmental delay: psychomotor delay, speech delay and intellectual disability, macrocephaly (p > 99) and obesity. Phenotype with facial dysmorphic features: wide forehead, deep set eyes, bulbous nose, prominent ears and pointed chin. The array-CGH analysis showed a 586 kb deletion at Xq21.1 including only one gene with associated disorder, BRWD3. Afterwards, the deletion was also identified in his asymptomatic mother and sister.
Conclusions. Our patient confirms that the haploinsufficiency due to BRWD3 deletion is a causal genetic mechanism of the BRWD3-related syndromic X-linked intellectual disability. It is important to recognize the phenotype for the diagnosis and follow up of the patients, and also to carry out the family genetic analysis in order to identify and give genetic counselling to the women who also have the genetic defect, because the majority of them are asymptomatic, as the mother and sister of our patient.
Key words. BRWD3 gene. Intellectual disability. Macrocephaly. Obesity. Overgrowth. X-linked.
|
© 2024 Revista de Neurología
 OPEN ACCESS
OPEN ACCESS

