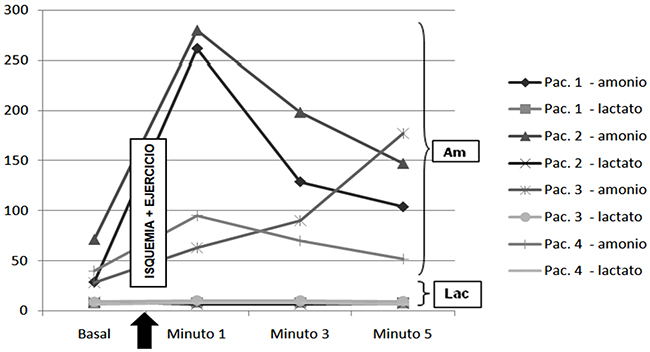
Tabla. Resultados de los cuatro pacientes.
|
| |
Paciente n.o 1
Varón, 9 años
|
Paciente n.o 2
Mujer, 14 años
|
Paciente n.o 3
Varón, 6 años
|
Paciente n.o 4
Varón, 9 años
|
Clínica inicial
|
Mucho cansancio y mialgias postejercicio
Second wind: dudoso (cinco años de evolución)
|
Dolor y debilidad al subir escaleras
Second wind: no (dos meses de evolución)
|
Mucho cansancio y mialgias postejercicio
Fue diagnosticado de ‘dolores de crecimiento’
Second wind: no (dos años de evolución)
|
Mucho cansancio y mialgias post-ejercicio
Second wind: sí (un año de evolución)
|
Orinas oscuras
|
No
|
No
|
Sí, con rabdomiólisis, sin insuficiencia renal
|
No
|
Exploración neurológica
|
Normal, con fuerza normal
|
Normal, con fuerza normal
|
Normal desde el tercer día tras la rabdomiólisis, con fuerza normal
|
Normal, con fuerza normal
|
Niveles de creatincinasa
(respecto al valor normal-alto = 288 UI/L)
|
× 3,5 (basal)
× 10 (tras el test isquemia)
Padre: creatincinasa × 30 (probable portador asintomático)
|
× 5 (basal, hacía 6 meses, aún asintomática)
× 16 (tras el test de isquemia)
|
× 18 (con tres años: en episodio de convulsión febril)
× 600 (en rabdomiólisis)
× 6 (basal)
× 16 (tras el test de isquemia)
|
× 2 (basal)
× 12 (tras el test de isquemia)
|
EMG
|
Miógeno
|
Miógeno
|
Miógeno
|
Miógeno
|
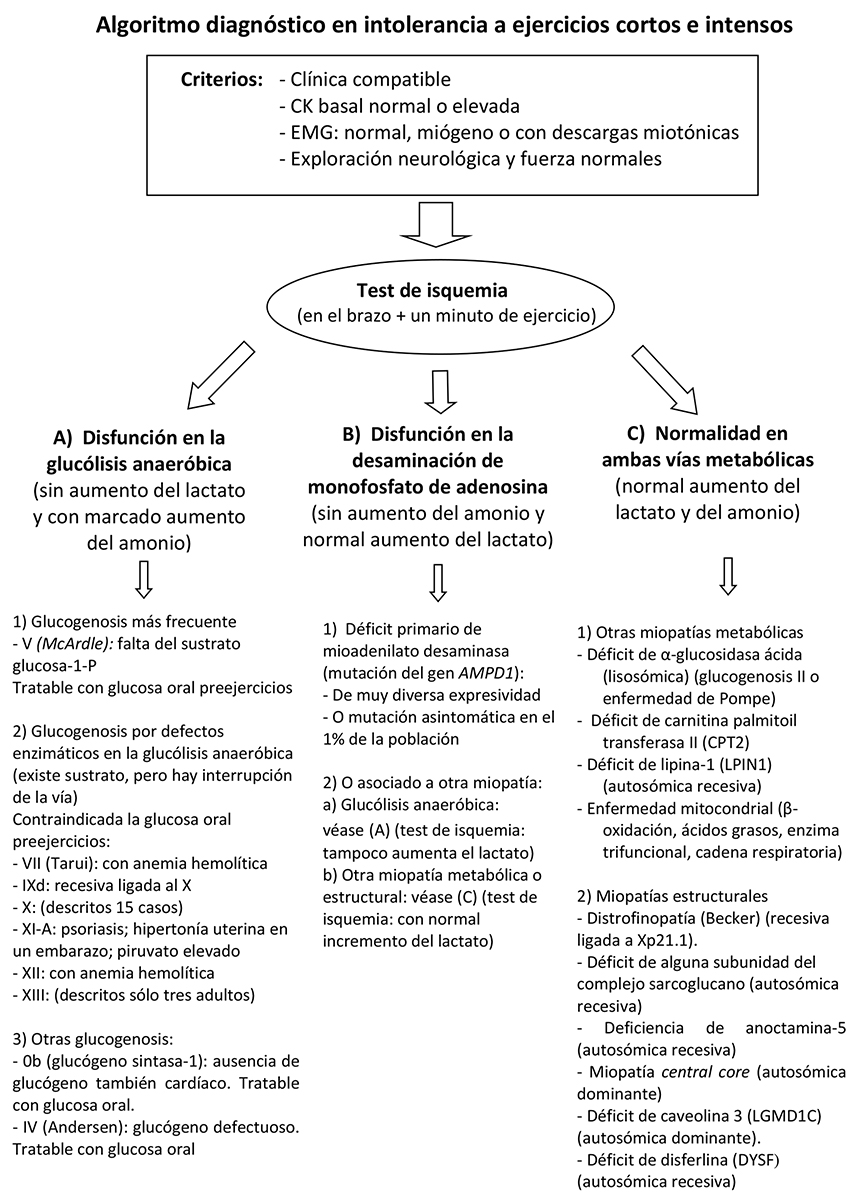
Test de isquemia
|
Disfunción en la vía de la glucólisis anaeróbica (contractura de 30 segundos en el brazo)
|
Disfunción en la vía de la glucólisis anaeróbica
|
Disfunción en la vía de la glucólisis anaeróbica
|
Disfunción en la vía de la glucólisis anaeróbica
|
Biopsia muscular
|
No practicada
|
No practicada
|
Acúmulos subsarcolémicos de glucógeno
Inmunohistoquímica: miofosforilasa negativa
|
No practicada
|
Gen PYGM
|
Mutación homocigota
c.2392T>C
|
Mutación homocigota
c.1963 G>A
|
Mutación homocigota
p.R50X
|
Mutación homocigota
p.R50X
|
 OPEN ACCESS
OPEN ACCESS